If you are citizen of an European Union member nation, you may not use this service unless you are at least 16 years old.
You already know Dokkio is an AI-powered assistant to organize & manage your digital files & messages. Very soon, Dokkio will support Outlook as well as One Drive. Check it out today!
DNA Methylation: What is it, and what does it do to us?
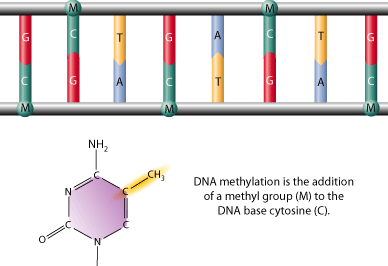
DNA methylation is a modifcation to DNA that occurs through the addition of a methyl group to DNA strand itself, often to the 5 carbon position of a cytosine ring. This type of modification is said to be an epigenetic modifcation that can be removed or inherited without any disruption of the original DNA sequence. Although DNA methylation is a common trait among vertebrates, it is more prevalent in some species than others. In addition, although DNA methylation is most common in normally unmethylated "CpG Islands (DNA regions where cytosine and guanine are located adjacently)," non CpG methylation has been documented in embryonic stem cells as well.
DNA methylation is thought to serve many purposes: It has been shown to be essential to normal development, X-chromosome activation, genetic imprinting, gene suppression, and carcinogenesis. Currently, our understanding of DNA methylation is limited, but there are a lot of people out there doing some really interesting work in efforts to understand what DNA methylation means to us as human beings (Do I have cancer? Am I at risk?), and how we can use this newfound understanding in applications that will help better our lives.
Understanding the Mechanism of DNA Methylation
What are CpG Islands?
As we know, DNA is made up of four nucleotide bases: Adenine, Thymidine, Cytosine, and Guanine. Adjacent nucleotide paings are sometimes referred to in a NpN fashion, where N is the base and p is the phosphodiester bond; thus, CpG is the designation given to Cytosine phospho Guanine. The frequency of the CpG dinucleotide is very low (or rather, sporadic) in the human genome. This is known as CG suppression. CG suppression is seen throughout our entire genome, except in small areas of 300-3000 bps long where the frequency of CpGs is either at the expected value or higher than expected. These areas are what are referred to as "CpG Islands," and represent roughly one percent of the genome. The theory as to why CpG islands escaped the phenomenon of CG suppression over evolution is because they are typically not methylated, and thus escaped mutational pressure.
CpG islands are most commonly found in the 5' region of expressed genes, and nearly 60% of human gene promoters are located in these regions. That is not to say that the islands are restricted to promoters of genes, as they may be found elsewhere in the genome as well. These purpose of these areas is still not fully understood.
Visual CpG island identification
CpG plots of the insulin like growth factor receptor genes from human (IGF2R, right) and mouse (Igf2r, left) were created by calculation of G+C and CpG content in sliding windows of 500 bp moved by 10 bp. CpG islands can be assigned visually as peaks of the CpG content (red) with at least 6 % CpG where also the G+C content (blue) exceeds 60 %. Thus, a prominent CpG island can be identified in the promoter region around the gene start, and two weaker ones are located inside of the gene.
Figure courtesy of the Center for Bioinformatics
What is DNA methylation?
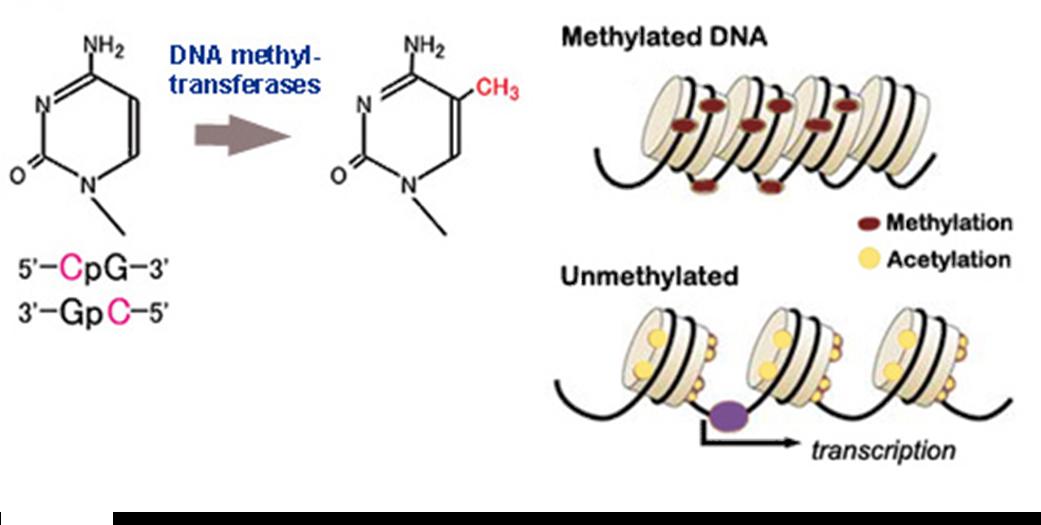
Methylation is an enzyme-mediated chemical modification that adds methyl (CH3) groups to specific locations on proteins, DNA, and RNA. DNA methylation only affects the Cytosine base (C) when it is followed by a Guanine (G). CpG sites in human DNA are methylated 70-80% of the time; however, methylation is mostly found in areas where CpG density is low, or at repetetive DNA sites. In healthy individuals, most CpG islands (where CpG density is high) are completely unmethylated.
As stated earlier, DNA methylation is invovled in many biological processes, but the function still remains incompletely understood. DNA methylation patterns are thought to be established during embryonic development, fixed, and then perpetuated through DNA-methyltransferase maintenance activity that recognizes and converts hemimethylated DNA to completely methylated DNA. The likelihood of DNA methylation at a given site has been recently suggested to be a dynamic process dependent on the holistic balance of all the factors constituting the mechanism machinery.
DNA methylation is understood to impact the transcription of genes in two ways. First, the binding of transcriptional proteins may be blocked physically just due to the the steric inhibition created by methylation. Secondly, the methylated DNA may become bound by methyl-CpG-binding domain proteins that recruit additional proteins, ending with the modification of the DNA into inactive chromatin ("silent chromatin"). Thus, there is a link between chromatin structure and DNA methylation. An example of the importance of this link is evident by the roles of methyl-CpG-binding protein 2 (MeCP2) and methyl-CpG-binding domain protein 2 (MBD2) in Rett Syndrome and cancer.
Plugin error: That plugin is not available.
Methylation primarily occurs at the C5 position of Cpg dinucleotides in mammals, and is characteristic of two general enzymatic activities: Maintenance methylation and de novo methylation. Some methylation is necessary in cells just for the preservation of existing DNA methylation following every ceullar replication cycle. Without this activity, unmethylated daughter strands would eventually be produced leading to passive demethylation. A deficiency in proteins essential for these maintenance activities was declared embryonically lethal after only 9 days in mice embryos (Haines et al., 2001). The process of establishing DNA methylation patterns during development is accomplished by a variety of de novo transferases (Doyle el al., 2002).
There has also been recent progress in the understanding of DNA methylation in plants. Differing from mammals, methylation occurs in plants in the CpG, CpNpG, and CpNpN context, where N represents anything other than G (Aufsatz et al., 200.
Proteins involved
DNA methylatin obviously requires some sort of machinery in order to function, and this methylation "machinery" includes multiple DNA-methyltransferase enzymes (some which have already been mentioned), de-methylases, methylation centers which trigger DNA methylation and are related topeculiar tertiary DNA structures, and methylation-protection centers that help keep CpG islands methylation free.
As previously mentioned, in order to avoid passive demethylation, DNA replication machinery needs to be equipped with DNA methyltransferase. DNMT1 is thought to be the maintenance methyltransferase believed to copy DNA methylation patterns to daughter DNA strands during replication. A deficiency of DNMT1 has been shown to be lethal to mice during development. It is also shown that inhibidition of DNMT1 alone might have potential for reactivating tumor suppressor genes silenced by DNA methylation. DNMT3a and DNMT3b are responsible for establishing DNA methylation patterns in early development. These have been referred to as de novo methyltransferases (Haines et al., 2002). DNMT3L is homologous to the other DMNT3 proteins but lacks catalytic acticity. It assists the other de novo transferases instead by increasing their ability to bind to DNA. DMNT2 is a protein established as a DNA methyltransferase homolog which contains all 10 sequience motifs constant to all DNA methyltransferases. Rather than methylate DNA, however, DNMT2 methylates cytosine-38 in the anticodon loop of aspartic acid transfer RNA.
CpG Island Methylation
CpG islands are thought to be normally barren of any DNA methylation. This is especially important for promoter-related CpG islands, because the un-methylated area is essential for transcription of the associated gene. The two important exceptions to this rule:
1) Genes on the inactive X-chromosome
2) Imprinted genes
In both situations, promoter-containing CpG islands are methylated at many CpG sites, and this methylation is associated with transcriptional repression of the gene. The triggers for CpG island methylation (in imprinted genes and on the X-chromosome) are still unknown but X-chromosome methylation occurs perhaps by allowing access of the islands to DNA-methyltransferase. Promoter-CpG island methylation is sufficient for gene silencing; continued presence is not necessary. On the X-chromosome, the trigger appears to be the RNA product of the Xist gene.
Initiation commences as methylated-DNA binding proteins are bound, followed by a protein complex that includes histones, deacetylases and other proteins. This protein complex then induces a closed chromatin structure that excludes transcription factors from the promoter area and results in gene silencing. When methylation is involved, silencing appears to be irreversible. When promoter CpG islands become methylated the gene associated becomes permanently silenced, and this silencing is transmitted through mitosis.
The first two genes that demonstrated CpG island promoter methylation in cancer were Calcitonin and MyoD. These findings lead to the hypothesis that promoter methylation may be one of the mechanisms of tumor-suppressor gene inactivation in cancer.
There are researchers out there who have attempted to put together lists of genes that have allegedly been affected by CpG island methylation (available here). The exact (or even approximate) number of genes hypermethylated in cancer is unknown. Equally as interesting are the genes which are not found to be methylated in aging or cancer, and there is also a sample list of those assembled on the MD Anderson Cancer Center's website here.
Our friends, the miRNAs and siRNAs
Keep in mind that there are other types of RNA out there, and it's important to have an idea of what they do before we get any further. As we know, the end result of gene expression is tied to translational control and mRNA degradation(Valencia-Sanchez and Hannon, 2006). It is now understood that small RNA molecules are modulators of gene expression in many eukaryotic cells. These small RNAs, capable of controlling gene expression, may either be endogenous or exogenous micro RNAs (miRNAs) and short interfering RNAs (siRNAs). Remember, siRNA is a class of 20-25 nucleotide-long double stranded RNA molecules that have a variety of functions. Perhaps most importantly, siRNA has been known to interfere with the expression specific genes. Additionaly, siRNAs also act in RNAi-related pathways such as anti-viral mechanisms, or in shaping the chromatin structure of a genome. Since these RNAs can affect mRNA degradation, translation, and chromatin structure, they are also capable of disrupting translation rates. For a pretty decent paper discussing possible mechanisms by which miRNAs control translation and mRNA degradation can be found here.
Methods used to study CpG Island DNA methylation
Methods for DNA methylation analysis can be divided roughly into two types: Global and gene-specific methylation analysis (MD Anderson). For global methylation analysis, there are methods which measure the overall level of methyl cytosines in genome such as chromatographic methods and methyl accepting capacity assays. For gene-specific methylation analysis, a large number of techniques have been developed (Dahl and Guldberg, 2003).
As with anything, the methods you should use to study CpG Island DNA methylation depend on what you are really interested in knowing. Basically, there are two types of experiments that you can do:
Distinguishing methylated from unmethylated DNA based on methylation-sensitive restriction enzymes
Distinguishing methylated from unmethylated DNA based on bisulfite conversion of DNA which, when completed, converts unmethylated C to U, but leaves methylated C intact.
Methylation-Sensitive Restriction Enzymes
Methylation-sensitive restriction enzyme digestion can be followed by Southern blot analysis, PCR, or newer techniques such as RLGS and MCA.
Some people believe that Southern blot methylation analyses is the best technique available if you need accurate quantitation of partial methylation patterns. This technique has been criticized because of the limited number of sites that can be analyzed. However, most Southern blot analysis are confirmed by more extensive analysis using bisulfite/sequencing. When using Southern blots, it is preferable to restrict with enzymes that have at least two CG sites in their recognition sequence (e.g. NotI, SacII, EagI, BssHII etc.) to increase sensitivity. The major downside of using Southern blots is that it is time-consuming and requires at least five µg of good-quality DNA.
Using methylation-sensitive restriction enzymes followed by PCR is tricky, as the technique is prone to give false positives due to incomplete digestion, and is particularly problematic if you span only one site. Bisulfite/PCR techniques will achieve the same results with a much lower rate of false positives. If this technique must be used, the best bet for success is to span multiple restriction enzyme sites, and use careful controls.
RLGS (restriction landmark genomic scanning) was developed primarily as a whole genome approach to methylation analysis, and to allow cloning differentially methylated sequences. It is too cumbersome to be used for the analysis of individual CpG islands.
MCA (methylated CpG island amplification) was developed as a high throughput method that is useful for analyzing large numbers of CpG islands in many samples relatively fast. It can also be used as a first step for cloning differentially methylated CpG islands. It requires relatively good quality DNA and is sensitive to partial digestion with restriction enzymes.
Bisulfite Conversion of DNA
Bisulfite conversion of DNA can be followed by several assays including sequencing, MSP, restriction analysis, Ms-SNuPE and others. All require PCR and are thus prone to quantitation problems inherent to PCR, as well as PCR bias, which is due to the fact that the methylated and unmethylated DNA molecules sometimes amplify with greatly differing efficiencies. However, all the techniques work on paraffin-embedded tissues and generally degraded DNA. False positives due to incomplete conversion can also be avoided by designing primers specific for the converted sequences.
Sequencing bisulfite converted DNA is the prime method for bisulfite techniques. Both direct sequencing and cloning PCR products followed by sequencing individual clones can be done. Direct sequencing is faster, but cloning resolves better issues of heterogeneity of methylation. Careful primer design is very important to avoid amplifying unconverted DNA (which will look heavily methylated at both CpG and non-CpG sites).
Methylation Sensitive PCR (MSP) is a rapid and very sensitive technique to screen for methylation: Primers are designed to amplify either the methylated strand or unmethylated strand, and a simple gel electrophoresis will yield the answer. MSP is the most sensitive technique available, and can detect 0.1 percent methylation relatively accurately. Its downside is a relative lack of quantitative information as well as occasional PCR problems.
Bisulfite PCR followed by restriction analysis has been described by several groups. After amplification, using primers that theoretically amplify both methylated and unmethylated strands, the PCR product mixture is digested with restriction enzymes that distinguish the two species and resolved using gel electrophoresis. This method is more time consuming than MSP due to an additional restriction step, but is more reliably quantitative. Still, PCR bias can result in serious quantitation errors (when using mixing experiments or comparing to Southern blots, for example).
Ms-SNuPE uses bisulfite/PCR followed by single-nucleotide primer extension, where incorporation of C (vs. T) denotes methylation. Results can be analyzed by dot-blot analysis, making it relatively fast and quantitative. Still, PCR bias might become an issue.
Obviously, there are new methods for analyzing DNA methylation being developed and tested, and some of them are actually successful. For example, in 2006, a patent was put out for a new method for high-throughput DNA methlation analysis received a patent. The patent, and all of the claims made regarding the process, can be found at http://www.patentstorm.us/patents/7112404/fulltext.html. Although it's a slightly rough read, I strongly recommend looking at it, just to appreciate the amount of work that has to go into a patent as well as establishing a quality scientific method.
How are Aging and DNA Methylation Related?
Commonly observed in aging cells is a global decrease in 5-methyl-cytosine levels (also indicitive of early neoplasia). The causes of this hypomethylation are not known. However, as CpG islands are not normally methylated, the processes of CpG island hypermethylation and global genomic hypomethylation are very different yet in fact still coexist in cancer and aging cells.
The Link Between DNA Methylation and Cancer
As we now know, epigenetic changes like DNA methylation help regulate gene expression in normal animal development. However, critical growth regulators such as tumor repressor genes may be subject to transcriptional silencing, thanks to hypermethlyation of specific promoter sequences (Baylin, 2005). Hypermethylation, which represses transcription of the promoter regions of tumor suppressor genes causing gene silencing, has been extensively studied in its relationship to carcinogenesis. In addition, it has also been shown that global hypermethylation may also cause oncogenesis by inducing chromosomal instability and spurious gene expression (Das and Signal, 2004; Ghoshal and Bai, 2007). There is an idea that dietary folate and methylene terahydrofolate reductase polymorphisms may contribute to varying methylation patterns, and the association of certain patterns to certain cancer tissues and normal tissues is still under study (see "Lung Cancer Diagnosis" below). On the up side, methylation occurs early in development and can be detedted in body fluids, providing the possibility of methylation patterns as diagnostic tools in detection and prognosis. Also, since DNA methylation is reversible, drugs like 5'-azacytidine, decitabine, and histone deacetylase inhibitors are being tested as treatments for a variety of different tumors. The field of DNA demethylation continues to expand with the onset of new demethylating agents such as antisense DNA methyltransferase and small interference RNA.
*Image courtesy of the cellscience website
The University of Texas MD Anderson Cancer Center has done some extensive research on the correlation between DNA methylation, aging, and cancer. Successful techniques have been developed to clone hypermethylated sequences in an effort to understand the potential imortance of CpG island methylation as a possible mechanism for the epigenetic changes associated with cancer and aging. Hopefully, these cloned sequences will lead to the identification of new genes which are important to cancer formation. Unfortunately, most of the sequences discovered don't correspond to currently known genes. The sequences cloned in the Anderson Cancer Center have been put online in an effort to collaborate with other labs. To see a list of their sequences, click here.
Another cool feature that the MD Anderson Cancer Center has included on their website is the ability to view not only a list of genes thought to be methylated in cancer, but also a list of genes that are not methylated in cancer. Links to both lists, as well as some other pretty nifty research, can be found at http://www.mdanderson.org/departments/methylation. As you may notice, the list of genes methylated in cancer is, sadly, quite a long list.
A complete rundown of cancer related facts and figures is provided in a report by the American Cancer Society, and may be viewed here.
The fundamental role of epigenetic events in cancer - This article is full of great information on DNA methylation in cancer, and epigenetics in general. The link isn't to the paper itself, but to the figures in the paper. I really recommend scrolling through them and reading the captions, as they are very informative. Here is illustration of one of the figures from the article:
DNA Methylation in the News: Current Research and Applications
Recently, an increased understanding behind the mechanics of DNA methylation has led to a diverse number of applications and uses in a variety of disciplines. In this section, we've highlighted a few interesting articles (to us) in an effort to introduce you to a wide range of topics related to DNA methylation in the news, new techniques, medical uses, and much more.
Using DNA Methylation as a Diagnostic Tool in Cancer Research
Colon Cancer Clue
New evidence reported by a team of researchers from the Whitehead Institute for Biomedical Research has indicated a possible link between DNA methylation and colon cancer, and may also provide a new method for treating this common cancer. Th current research focuses on familial adenomatous polyposis, an inherited form of colon tumor disease, which may afflict people as early as the age of 40. Benign polyps are a signature of the disease, and patients may develop as many as 1,000, which, if not surgically removed, may progress into colon cancer. Mice, given an equivalent mutation in the gene responsible for FAP, provided an ideal system for studying the origins of tumors in humans. These mice, called Min mice, were interbred with a strain of mice that had an induced shortage of the enzyme DNA methyltransferase. This enzyme of responsible for adding methyl groups to DNA. Interestingly enough, the interbred mice had 60% fewer polyps after 180 days than the control Min mice.
The researchers where also able to reduce the formation of precancerous tumors in the mouse models with a preventative drug treatment intended to inhibit DNA methylation. When a drug known to inhibit DNA methyltransferase, 5-aza-2'-deoxycytidine (5-aza-dC), was used to further reduce enzyme levels in the test mice, a 98% decrease in polyps was seen. An 80% decrease in polyp formation was seen in control Min mice with just the 5-aza-dC treatment alone. Although the treatment is currently too toxic to be useful as a preventative treatment in humans, it indicate that there are a lot of possibilities out there for creating a drug that might be safe for humans, and a possible colon cancer preventative. The next goal of the researchers is to explore whether or not it's possible to safely inhibit DNA methyltransferase activity in cells lining the human colon, with the long term goal being that of suppressing polyp formation in patients already diagnosed with FAP. According to the American Cancer Society, colon and rectum cancer is responsible for 8% of cancer deaths in males and 9% of cancer deaths in females. Ten percent of new cancer cases are attributed to colon and rectum cancer in both males and females.
In another related study, researchers report an assessment of faecal DNA from patients afflicted with colorectal cancer and controls to determine the "feasability, sensitivity, and specificity" of this approach (Muller et al., 2004). SFRP2 methylation was used as a single DNA-based marker for identification of colorectal cancer in stool samples from three independent sets of patients using a MethyLight analysis. To learn more about the possible use of genetic and epigenetic markers in colorectal cancer identification, view the complete article here.
For a list of articles related to colorectal cancer screening based on DNA methylation, click here.
Lung Cancer Diagnosis
Currently, lung cancer (carcinoma) is the leading cause of cancer death in the United States, as well as worldwide. Since the survival rate for cancers of this type of so depressed (6-16%, depending on cell type), the best chance in improving survivorship rates lies in early detection. Although there are groups of people more likely to develop this cancer, screening of likely candidates, especially long-term smokers, has not been successful in reducing lung cancer mortalities. The identification of lung cancer-specific biomarkers, and non-invasive methods for the detection of these biomarkers at an early age, may be promising in the effort to identify cancers. It has previously been demonstrated that hypermethylation of CpG islands in the promoter regions of genes is a common occurence in lung cancer. This has been demonstrated through the analysis of the methlation status of over 40 genes associated with lung cancer tumors and cell lines. Specifically, it has been noted that DNA methylation patterns are significantly altered in cancerous cells (Jones and Laird, 1999; Robertson, 2001). A genome-wide hypomethylation is generally observed in cancer cells. This may lead to carcinogenesis through oncogene activation, retransposon activation, and chromosome instability. Concurrently, hpermethylation of certain CpG islands is seen, which may be associated with gene silencing. The methylated residues are believed to recruit methyl-binding proteins (and associated factors), thus leading to transcriptional shut-down and chromatin remodeling. Preliminary studies has demonstrated genome-wide and gene-specific hypomethylation in lung tumors compared to normal tissue counterparts (Feinberg and Vogelstein, 1983). Several possible mechanisms has been proposed to account for hypomethylation's role in tumorigenesis. Eventually, the goal is to understand these patterns better and to develop non-invasive techniques for detecting genetic biomarkers. High specificity is a must if these biomarkers are going to be used as a diagnostic tool, and the aprehension that false positives might be obtained from some individuals exists as well. For a more complete review of the study, the original article is available for viewing here.
DNA Methylation and Demethylating Drugs in Myelodysplastic Syndromes and Secondary Leukemias
Article: Inhibitors of DNA methylation in the treatment of hematological malignancies and MDS. Author: Leone et al.
Abnormalities in DNA methylation have been identified as one of the most common molecular changes found in hematopoetic neoplasms (tumors). Hematopoetic neoplasms, as defined by the National Cancer Institute, are those specifically located in the blood and blood-forming tissues (bone marrow and lymphatic tissues). Although we've already established that hypermethylation of genes involved in cell-cycle control and apoptosis most likely play a role in the development of cancer, in particular, high-risk myelodysplastic syndromes (MDS) and secondary leukemias show a particular high prevalence of tumor suppressor gene hypermethylation. An increase in methylation rate is linked to the progression of MDS, lymphoproliferative diseases, and myeloproliferative diseases, suggesting a link between hypermethylation of cirtical promoter regions in the progression to more aggressive phenotypes. However, irreversible DNA methyltransferase inhibitors like Decitabine and 5-azacytidine provide hope for the treatment of MDS and acute myeloid leukemia. A sequential administration of a "first generation" demethylating agent and HDAC inhibitors showed the potential for reducing methylation of target genes. Hopefully, the results of clinical trials will show that the natural history of MDS may be altered using a non-invasive threatment, characterized by an outstanding toxicity profile.
In a similar article, DNA hypermethylation of myeloid cells, a nobel therapeutic terget in MDS and AML, the authors also examine the potential of DNA demethylation of target genes using DNA methyltransferase inhibitors. This study focuses a bit more on the karyotypes of patients, as well as specific abnormalities.
Difficulties with Finding a Good Drug
Although clinical trials using Vidaza (5-azacytidine) and Dacogen (5'aza-2'-deoxycytidine) have been quite successful against leukemia such as MDS, acute myeloid leukemia, chronic myelogenous leukemia, and chronic myelomonocytic leukemia, their effectiveness against solid tumors has been less positive (Ghoshal and Bai, 2007). This has given researchers the goal of developing new inhibitors that are less toxic and operatie with more efficacy. The downfall of these drugs as anticancer agents is their instability in vivo along with secondary toxicity leading to cell cycle arrest due to their excessive incorporation into DNA. Therefore, the treatment of cancer cells with stable drugs that are capable of facilitating gene activation in cancer cells through the increaed degradation of DNA methyltransferases without DNA incorporation would be the ideal route.
Miscellaneous Research Concerning DNA Methylation
Global Hypomethylation and Chromosomal Instability Linked with Ovarian Cancer - What do Folate, methionone, and B6 have to do with this?
Article: Intake of Folate and Related Nutrients in Relation to Risk of Epethelial Ovarian Cancer Authors: Tworoger et al.
Beginning in 1976, research has been conducted in order to help determine what factors may increase the risk of ovarian cancer in women. It was determined in the 22 year follow-up that there were no correlations between total folate, dietary or total vitamin B6, or methionone. A modestly decreased risk was associated with higher dietary folate after the exclusion of certain factors. In short, there was little evidence to believe that folate, methionone, and B6 were related to ovarian cancer risks.
Arabidpsis thaliana: New pathways that exclude cytosine methylation from genic regions, and regruitment of non-CG DNA methylation and initiation of siRNA spreading
Differential cytosine methylation of repeats and genes is essentialy for good genome stability and the proper expression of genes. A jmjC-domain gene, IBM1, has been identified in Arabidopsis thaliana through a genetic screen of mutants that showed exctopic cytosine methylation in a specific genic region (Saze et al., 2008). The ibm1 mutations also created a range of developmental phenotypes that depended on methylation of histone H3 at lysine 9. These phenotypes were oddly enhanced by the mutation in the chromatin-remodeling gene DDM1, a gene necessary for keeping methylation and silencing of repreated heterochromatin loci. In particular, this study focuses on the genomes of plants. Plant genomes (and others as well) include a large amount of transposons and DNA repeats, sequences that are potentially deleterious and cytosine-methylated in order to form heterochromatin (inactivated). Methylated heterochromatin has the potential to spread to flanking cellular genes and screw up their expression, especially in gene-rich regions. Mechanisms for confining methylated regions have remained elusive so far, despite their importance. In this study, a new pathway was identified in plants that excludes cytosine methylation from genic regions thanks to histone modification and chromatin remodeling, and appears to aid in proper plant development.
Further study of A. thaliana proved to be even more insightful. Apparently, plants use siRNAs to target cytosine DNA methylation to both symmetrical CG sequences and nonsymmetrical (CHG and CHH) sequences (Henderson and Jacobsen, 2008). The reason for using A. thaliana is that siRNA and DNA methylation clusters most commonly overlap with transposons in this plants' genome. The loss of the majority of non-CG DNA methylation in three different mutants led to varying developmental phenotypes (drm1, drm2, cmt3). The gene responsible for the different phenotypes was identified by the researchers as SDC (supressor of drm1 drm2 cmt3). SDC possesses seven promoter tandem repreats and also encodes for an F-box protein. According to the study, the "SDC repeats show a unique silencing requirement for non-CG DNA methylation directed redundantly by histone methylation and siRNAs." They also demonstrate the ability of siRNAs and methylation to spread beyond the repeated region. This study shows that plants, especially A. thaliana, can have important implications for understanding how plants utilize gene silencing, and these studies show overall how much we have yet to learn about the elusive plant genome.
Miscellaneous Fun? Stuff
Epigenetics in Print
If you're interested in the epigenetics aspect of DNA methylation, epigenetics in general, or in the mammalian genome, there is full access to a very interesting book, completely available online, that might pique your interest. The book is titled Epigenetics by Jorg Tost. The goal of the editor was to assemble a group of top-notch scientists from a wide spectrum of epigenetics related fields in order to create a new volume just on current epigenetics research. This volume gets into detail concerning the molecular mechanisms and biological processes in which epigenetic modifications play a role. "This up-to-date volume is a major resource for those working in the field and will stimulate readers of all levels to dive into the fascinating and fast moving field of epigenetics."
Wonderful outline. We'll be covering some of this in class so your treatment here will be a great complement to the course. Make sure to indicate who is doing what on the page. Nice Job..ck
You don't have permission to comment on this page.
This Sidebar appears everywhere on your wiki. Add to it whatever you like -- a navigation section, a link to your favorite web sites, or anything else.


 Plugin error: That plugin is not available.
Plugin error: That plugin is not available.






{kind=link}
{kind=link}
Comments (1)
Christopher Korey said
at 1:17 pm on Oct 15, 2008
Wonderful outline. We'll be covering some of this in class so your treatment here will be a great complement to the course. Make sure to indicate who is doing what on the page. Nice Job..ck
You don't have permission to comment on this page.