Klinefelter's Syndrome
Lani Van de Poel and Timothy Legare
Introduction
Originally identified by Dr. Harry Klinefelter in 1942, Klinefelter's syndrome is a sex chromosome condition that most commonly produces the XXY genotype in males. Other Karyotypes can be seen but are much less common such as the XXXY, causing even greater negative phenotypic effects. It is associated with many defects and symptoms that cause infertility, disfigurement, and many emotional and pyschological problems. The syndrome is typically found in 1 of every 500-1000 male births, and isassociated with little or no pubic hair, small penis, and the expression of many female features in the male patient. It can also induce many autoimmune diseases such as Lupus and certain cancers such as breast cancer and the developement of extragonadal tumor cells. More information on the basics of Klinefelter's syndrome can be found at genome.gov and the KS&A site
Causes:
The overlying cause of Klinefelter's Syndrome (KS) is the presence of one or more X chromosomes, or a mosaic of XY/XXY cells. The presence of this extra chromosome is due to nondisjunction during the formation of the egg or sperm cell. Nondisjunction can occur in meiosis I or II, when chromosomes separate unevenly at the poles, resulting in both n+1 and n-1 gametes. When an n+1 gamete fuses with an n gamete, a trisomic 2n+1 zygote is produced (Griffiths et al. 2000). In Klinefelter's, either an XX egg fuses with a Y sperm or an X egg fuses with an XY sperm depending on the parental origin. Nondisjuction occurs randomly with equal chance of maternal origin and paternal origin. These particular fusions of the egg and sperm have been linked to the failure of crossing over to occur. This usually happens because the recombinant sections of the chromatin are either too close or too far from the centromere. Another conjecture for the sex chromosome aneuploidy is that during meiosis I, recombination is either absent or aberrant. Hall et. al (2006) gathered data from multiple studies and found that over 70% of the 47, XXY males studied resulted from this error in meiosis I. This is a particularly high percentage and gives insight into how these chromosomal abnormalities arise. There is an increased risk of nondisjunction occuring in parents past a certain age, usually in mothers over 35 and fathers over 45. In mothers, it is thought that biochemical changes to the environment of primary oocytes occurs over time, reducing the efficiency of chromosome pairing in meiosis (Parsons 1964). Similarly, with increased paternal age, there is reduced efficiency in chromosome pairing during spermatogenesis. Other causes of nondisjunction stem from radiation or chemical exposure, viruses, or effects of malnutrition (Burson & Barringer 1998). Due to the randomness of nondisjunction causing KS, it is not based on an inherited factor and does not run in families.
Animation Of Meiotic Nondisjunction
Genetic Basis for KS Phenotypic Variability:
Normally, when two X chromosomes are present, one X is inactivated. It has been shown that there is preferential inactivation of one X chromosome over the other within an individual (Iitsuka et al. 2000), possibly leading to skewed X chromosome expression that could explain the wide range of phenotypes observed. It is suggested that if the maternal X is inactivated, all expressed genes will come from the father, which could lead to more severe symptoms (Wilkstrom et al. 2006). Likewise, if the paternal X is inactivated, the individual will have a more normal phenotype, as it will have gene expression more similar to a normal male with a maternal X chromosome and a paternal Y chromosome. If both X's are of maternal origin, and are identical due to an error in either meiosis II or mitosis, there could be expression of recessive mutations that could lead to more severe symptoms. It would seem that due to X inactivation, no such recessive mutations would be expressed, but in actuality, about 15% of the genes on the inactive X are still expressed. This results in over expression of some of the X chromosome genes, which is the main cause for many of the observed symptoms. With a greater number of X chromosomes, gene expression increases and symptoms are more severe.
Gene Expression in the XXY:
Recent studies have attempted to identify the genes that are over expressed and escape normal inactivation. Vawter et al. (2006) showed dysregulation of the X-linked genes and found that there were 129 diferentially expressed genes(DEGs) in the KS group compared to the XY controls. 14 of these DEG's were located on the X chromosomes and the other 115 located on autosomes. None of the differentially expressed genes were located on the single Y chromosome. The 14 X-linked genes that were differentially expressed were: XIST, MBTPS2, CXorf21, ZNF275, DSYS155E, SMCX, ZFX, MECP2, RPS4X, TAF9L, GTPBP6, SLC35A2, YY2, PGK1. This table shows the results for the amount of expression of these genes using two methods, microarray analysis and QPCR. Both methods returned similar results, with XIST having the most change in expression in XXY individuals compared with XY individuals.
XIST- The XIST gene is especially important, because it is essential in the X-inactivation of XXY individuals. When the XIST gene undergoes transcription, non-coding RNA is produced which causes the inactivation of other genes on the inactive X (Gilbert & Sharp 1999). The XIST gene is only transcribed on the inactive X. Gilbert & Sharp (1999) found that while both the active and inactive X chromosomes have histone acetylation at some gene promoters, many of the promoters on the inactive X are hypoacetylated, leading to tightening of the chromatin structure and lowering the chance that transcription factors can access the promoter and begin transcription of the gene. This hypoacetylation, along with the methylation of CG's on the inactive X, are key in regulating the inactivation of the X chromosome. When the genes on the inactive X are methylated or hypoacetylated, they cannot be transcribed, and therefore cannot be translated into proteins. When they escape inactivation, they are able to be transcribed and translated and ultimately twice the amount of the translated protein (one from each X chromosome) is produced, which is called overexpression of the gene.
Androgen receptor gene- The androgen receptor gene is responsible for the regulation of testosterone and 5alpha-dihydrotestosterone(DHT) that leads to the production of internal and external male genitalia. The AR gene codes for a ligand-dependent transcription factor. The N terminal domain of the first exon of the gene contains highly polymorphic repeats of CAG, and when translated into a protein, these repeats create the N- terminal transactivation domain of the protein. The number of repeats determines the activity of the gene receptor(Zinn et al. 2005). Longer CAG repeats have been associated with underdeveloped genitalia(Lim et al. 2000). Men with KS have two X chromosomes, and therefore two AR genes, but in every cell, one X is inactivated and the AR gene is methylated so that it cannot be transcribed. Zitzmann et al. (2004) showed that the length of CAG repeat(CAGn) was positively correlated to body height, and negatively correlated to bone density and the relation of arm span to body height. When CAGn was short, the receptor gene was preferentially inactive, but a long CAGn was predictive of small testes and the development of male breasts, or gynecomastia. They also found that there was a preferential inactivation of the X chromosome with the longer CAGn on the AR gene, as seen in the figure below. Behaviorally, those with longer CAGn had a lesser chance of being in stable relationships and overall had less professional achievements.

Figure1:
Zitzmann et al. (2004) show here in A the correlation of short and long alleles for the CAGn to inactivation, with short alleles inactivated more often. In B, they show the difference between the X-weighted mean and the arithmetic mean for length in CAGn alleles in men with both alleles present . The difference is seen to be above zero, showing that the average expressed length is longer than just the average of all possible lengths. In the example, the triangle represents a short CAGn of 20bp, the diamond an arithmetic mean of 25 bp, the dot a longer X-weighted mean of 27bp, and the square a long CAGn of 30bp.
SHOX gene- The SHOX gene, located on the PAR1 pseudoautosomal region (seen in the yellow bracket of this image) of the X chromosome that escapes X inactivation, is the short stature homeobox containing gene.It is also found on the Y chromosome, so in KS men, there are three copies of the gene undergoing transcription, and therefore three copies of the translated protein present. It is primarily expressed in bone marrow fibroblasts, especially in the distal limbs. It is thought that the proteins translated from the SHOX gene transcribe act as repressors for the fusion of growth plates and prevent skeletal maturation, leading to the tall stature, largely due to long limbs, found in the common KS phenotype(Ogata et al. 2001).
Diagnosis:
Due to the wide variety of phenotypes resulting from KS, only about 25% of expected cases are diagnosed, most of which are already in adulthood (Bojesen et al. 2004). Many reasons are brought up for why this diagnosis rate is so low including that karyotyping is not common amongst newborn babies, prenatal genetic tests are invasive, and also that many cases of Klinefelter's syndrome are now beleived to be intrauterine deaths (Bojesen and Gravholt 2007). Intrauterine death means that the developing fetus dies while in utero. This is important because it skews the actual amount of cases and leads researchers to beleive that this disorder is not as common as it may really be. Most symptoms are first seen at puberty when androgen deficiency becomes evident as testes begin to shrink and harden, gynecomastia develops, and accelerated growth is observed. In adults, diagnosis is most common when infertility is combined with common KS phenotypic traits. Presently, diagnosis still relies on karyotyping, but this method is time consuming and can only be performed in specialized labs (Ottesen et al. 2007). Early diagnosis is of utmost importance for KS patients because early treatments could prevent many of the long-term effects experienced, even infertility. Onay et al. (2008) present the use of quantitative fluorescent polymerase chain reaction (QF-PCR) of amniotic fluid as an efficient prenatal diagnostic method of testing for aneuploidy. Their method gave results with a few hours, was low in cost, and gave results consistent with cytogenetic studies. Ottesen et al. (2007) present a similarly simple method of diagnosis, using quantitative real-time PCR (qPCR) analysis of androgen receptor gene copy number. They found this method to be specific and reliable, and usable for screening newborns. These screening methods take less time and money than conventional karyotyping, so their use will help in early diagnosis of KS, leading to early treatment and prevention of long-term negative effects.
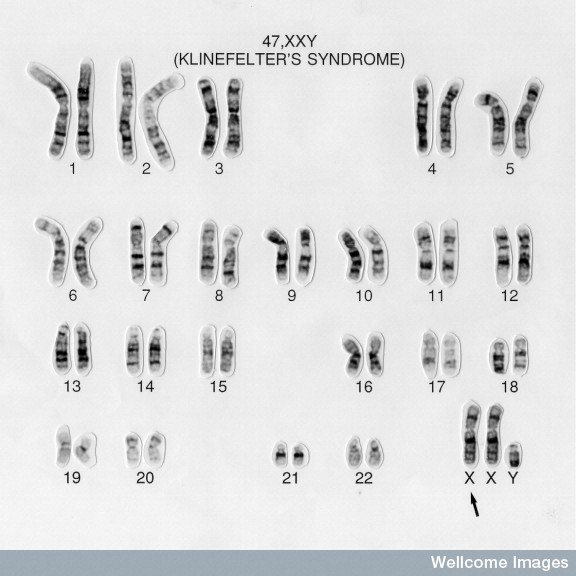
This image shows a karyotype of an idividual with KS. Karyotyping is the most accurate and commonly used diagnosis tool for KS. The arrow is pointing at the extra X chromosome.
Treatments:
The main treatment for Klinefelter's is testosterone supplementation in order to counteract the effects of female expressing genes in the body. If diagnosis is caught early, around 11-12 years old, then the prescribed doses of testerone may vary, but the expected results will be
- Increases body hair, mainly on the face (beard), under the arm (axillary), and in the genital area (pubic).
- Increases muscle development.
- Increases sex drive.
- Helps prevent osteoporosis
- May prevent or shrink enlarged breasts.
- Provides better self-esteem by allowing the boy to "fit in" with his peers. This can result in more successful interpersonal relationships.
All these effects are directly resultant of the addition of testosterone into the pre/pubusecent body. Depo-testoterone ( the man made form) may be injected into the body through a hypodermic needle, or it can be absorbed through the skin in patch form. As aging continues the patient may develop learning disabilities that can be coached through extra educational services and the testoterone treatments will continue. This hormonal treatment does not reintroduce fertility into the male, but certainly helps with the phenotypic results of this defect (WebMD article.) Along with the testosterone treatments, physcians will prescibe additional hormal treatments to counteract the phenotypic symptoms of Klinefelter's. Some of the most common that the doctors will give along with testoterone are: FSH, follicle stimulating hormone to also help increase pubic hair growth and body hair, LH, which is leutinizing hormone a hormone produced in the anterior pituitary gland that promotes the leydig cells in the testes to produce testoterone, and estradiol which is a form of estrogen believed to prevent the apoptosis of male germ cells. (Visootsak & Graham 2006) All of these hormonal treatments combined generally help to reduce the effects of Klinefelter's but mainly only the phenotypes not the infertility man usually must deal with. Also these treatments are given in increasing amounts as puberty progresses as normal, in order to not overload the body.
Symptoms
Symptoms are generally caused by the overexpression of genes coded on the X chromosome. The most common symptom seen in men with this genotype is infertility, but many other symptoms can be seen such as sexual problems, small testicles and penis, unusual body proportions, and gynecomastia. These are the most common symptoms of Klinefelters, but other autoimmune dieases can arise from the complications seen in the XXY genotype. (NYTimes).
The following video goes over the basic symptoms found in KS individuals:

Complications
Many complications can arise from the Klinefelter syndrome and other Sex chromosome aneuplodies. One recently studied complication that is associated with sex chromosome aneuplodies is the development of intercranial germ cell tumors. IGCT tumorigenesis had been linked to Klinefelter's by researchers at the General Hospital of Mexico ( Quepo et al. 2008). This was only performed as a statistical analysis, because the pathway to the creation of these malignant tumors is currently unknown. It has been proclaimed that this constitutive anueploidy is linked to carcinogenesis. Another result of sexual aneuploidy is the defects it can cause in the brain. Due to the influence of the aneuploidy the brain XXY phenotypes develops to be much smaller in comparison with normal genotype individuals. In particular the Gray Matter of the brain in XXY patients is 8.5% less than that seen in normal genotypic patients. Also the White Matter of the brain is reduced in size by an 8.1% comparison decrease. In addition to the decreases in Gray and White Matter of the brain XXY individuals had a significant (47% in comparison) increase of cerebrospinal fluid of the brain. Cerebrospinal fluid is the fluid that resides in and around the brain. It is the fluid on which the brain rests inside the skull (Giedd et al. 2006). These deficiencies induce congitive defects that are the most common amongst men with sexual anuepliody (Klinefelter's Syndrome) and women (Turner's Syndrome). The women have spatial problems in the development of the brain whilst the men in this study have trouble trying to acquire words for speech. These problems have been linked with the aneuploidy because TS women have an increased leftward brain asymmetry particularly superior temporal and parietal-occipital association complex where as the men portrayed throughout the frontal lobes. These results show that the number of sex chromosomes in patients influences the brain asymmetry. All these results were found by researchers at the University of Liverpool (Rezaie et al. 2008). Another main concern in individuals with sex chromosome aneuploidy is the development of breast cancer in men. Male breast cancer is a very uncommon condition, accounting for only 0.2 percent of all male cancer patients, but Klinefelter's patients have a much higher risk. Approximately 6% of all Klinefelter's patients develop breast cancer at some point in their lives, but all patients have a 20 fold increased risk of developing breast cancer depending on diet and other such choices as smoking (Hsing et al. 1998)
Connection to the Central Dogma:
Klinefelter's Syndrome is a sex chromosome aneuploidy that is caused by nondisjunction of chromatids in either meiosis I or II, so transcription and translation are not involved in the cause of this genetic disorder. In particular this nondisjunction seems to be completely random, but can be induced in cells to make this occur. Certain environmental effects can cause this nondisjunction to occur, such as the chemical influence of alkylating agents and antimetabolites inducing nondisjunction to occur in mice. The mechanisms of how these agents induce the nondisjunction are not completely elucidated yet, and further study must be performed to determine how these chemicals cause the nondisjunction. The phenomenon has also been shown to be induceable by using x-ray irradiation. The x-rays cause significant chromosome damage and change through a free radical mechanism where one electron is being cleaved off of atoms causing their reactivity to greatly increase(Sgura et al. 2001 & Hansmann & Probeck 1979). While these inductions of nondisjunction are helpful in learning more about the occurences of nondisjunction, they leave a great hole of information in how the body produces these karyotypes randomly in nature. A few conjectures have been raised about the reasoning behind nondisjuction in meiosis that occurs in nature. Firstly, a failure to recombine in the homologous chromatin could be the reason for this happening in man. Because the recombinant sections of DNA are either too far or too close to the centromere, recombination does not occur and the failure to segregate in anaphase produces the aneuploidy of the sex chromosomes. Another conjecture that was discovered was the influence of the structural differences that occur between the fibrillar centers on the chromatid and whether or not these centers contain a certain sequence of rDNA. If during crossing over of nonhomologous chromosomes occurs on the outer limit of the fibrillar center, it produces a dicentric chromosome lacking these ribosomal genes. Conversely, if the crossing over occurs deep in the fibrillar center, a dicentric chromosome will be produced with these ribosomal genes. Both situations that were studied in mice produced higher incidences of nondisjunction and chromosome damage (Mirre et al. 1980). These studies show that more experiments need to be designed to find the reason for this phenomenon of nondisjunction, especially the roles of the ribosomal DNA and the fibrillar centers and spindle proteins. The connections of KS to the central dogma lie in the overexpression of certain genes. When genes on the inactive X chromosome either escape inactivation or are hyperacetylated, they will undergo transcription into RNA. This RNA will either be non-coding, such as for the XIST gene transcribe, or coding, such as for the SHOX gene. The coding RNA will undergo translation and code for a specific protein to be made.
Works Cited
{kind=link}
{kind=link}
{kind=link}
Comments (1)
Christopher Korey said
at 1:36 pm on Oct 15, 2008
Looks good. An in depth coverage of the molecular consequences (gene expression etc.) of sex chromosome abnormalities will be a great addition to the page. Make sure to cover more of the molecular details than the symptoms and treatments. Nice Job!..ck
You don't have permission to comment on this page.